扩增子测序是对特定长度的PCR产物或者捕获的片段进行测序。16S/18S/ITS等扩增子测序即通过提取环境样品的DNA,选择合适的通用引物扩增16S/18S/ITS的某一或某几个区,使用Illumina MiSeq/HiSeq将目的区域正反向读通,通过检测目的区域的序列变异和丰度,对环境样本物种分类及丰度、种群结构、系统进化、群落比较等方面信息进行分析的研究方法。
宏基因组 ( Metagenome)(也称微生物环境基因组Microbial Environmental Genome, 或元基因组)是由 Handelsman 等 1998 年提出的新名词, 其定义为“the genomes of the total microbiota found in nature” , 即环境中全部微小生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因,目前主要指环境样品中的细菌和真菌的基因组总和。宏基因组学(或元基因组学,metagenomics)是一种以环境样品中的微生物群体基因组为研究对象,以功能基因筛选和/或测序分析为研究手段,以微生物多样性、种群结构、进化关系、功能活性、相互协作关系及与环境之间的关系为研究目的的新的微生物研究方法。一般包括从环境样品中提取基因组DNA, 进行高通量测序分析,或克隆DNA到合适的载体,导入宿主菌体,筛选目的转化子等工作。
特定生物种基因组研究使人们的认识单元实现了从单一基因到基因集合的转变,宏基因组研究将使人们摆脱物种界限,揭示更高更复杂层次上的生命运动规律。在目前的基因结构功能认识和基因操作技术背景下,细菌宏基因组成为研究和开发的主要对象。细菌宏基因组、细菌人工染色体文库筛选和基因系统学分析使研究者能更有效地开发细菌基因资源,更深入地洞察细菌多样性。
全基因组测序分为从头测序(de novo sequencing)和重测序(re-sequencing)。
从头测序(de novo)不需要任何参考基因组信息即可对某个物种的基因组进行测序,利用生物信息学分析方法进行拼接、组装,获得该物种的基因组序列图谱,从而推进该物种的后续研究。
基因组重测序是对有参考基因组物种的不同个体进行的基因组测序,并在此基础上对个体或群体进行差异性分析。基因组重测序主要用于辅助研究者发现单核苷酸多态性位点(SNPs)、拷贝数变异(CNV)、插入/缺失(Indel)等变异类型,以较低的价格将单个参考基因组信息扩增为生物群体的遗传特征。全基因组重测序在人类疾病和动植物育种研究中广泛应用。
微生物是地球上最丰富、最多样的生命形式,它们是所有生态系统的重要组成部分,在医疗健康领域和工业应用领域中发挥着至关重要的作用。微生物基因组学研究在医学上可应用于致病相关基因的鉴定、开发新型抗生素等,而在生物技术上可用于生物降解、酶工业、食品生物以及抗生物质的研究,同时对进化、功能预测等方面也有着重要意义。
自1994年美国发起微生物基因组研究计划(MGP)以来,微生物组学在生物领域蓬勃发展,而在2001年中国绘制出首张微生物基因组“完成图”以后,随着高通量测序技术的诞生,微生物基因组研究也进入了井喷期,我国先后启动了万种微生物基因组计划、百万微生态系统基因组计划等。
研究的深入依赖技术的进步。短读长测序技术由于无法跨越富含重复区域的基因组使得很多微生物研究止步于基因组草图,而长读长测序技术却可以轻松获取细菌基因组完成图,同时在复杂微生物群落的鉴定中可达到“种级别”的鉴定水平。Frank等人于2016年结合Hiseq2000和PacBio RS II对沼气反应器内微生物宏基因组进行研究发现PacBio技术能显著提升组装水平,而2013年Chin等仅运用PacBio就组装出16个微生物基因组,与参考基因组一致性达99.9999%。
如果说PacBio技术为微生物研究打开新世界的大门,那么基于纳米孔电流检测的ONT技术则开启了微生物组学的黄金时代。

我们拥有专业的生信分析团队,除常规的基础分析、标准分析外,可根据您的需求,个性化定制分析方案、优化分析流程,解除您的后顾之忧……
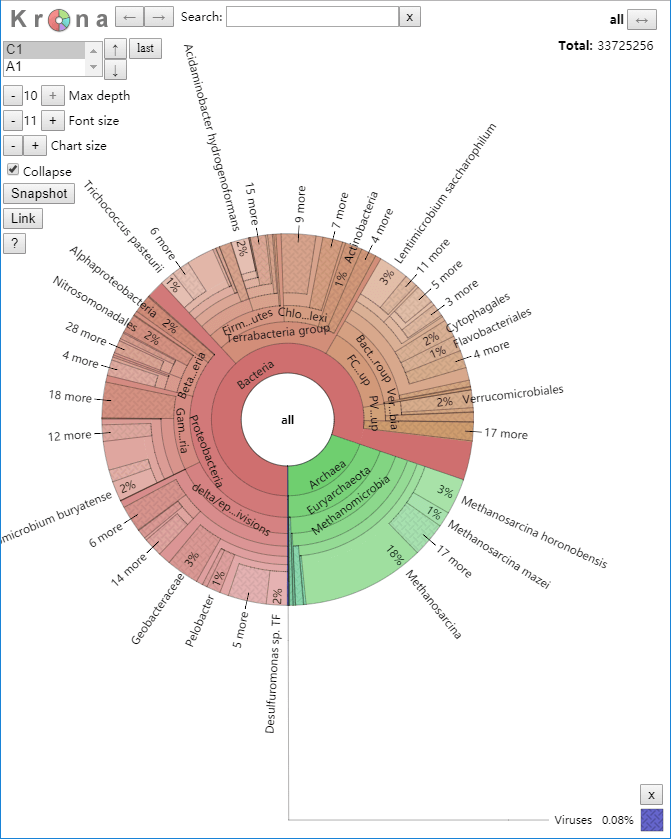
Krona分析,通过可视化交互形式,直观展示不同样本或分组之间的物种多样性的差异。
线性判别分析(Linear Determinant Analysis, LDA)是对费舍尔的线性鉴别方法的归纳,这种方法使用统计学、模式识别和机器学习方法,试图找到两类物体或事件的特征的一个线性组合,以能够特征化或区分它们。所得的组合可用来作为一个线性分类器,或者更常见的是,为后续的分类做降维处理。
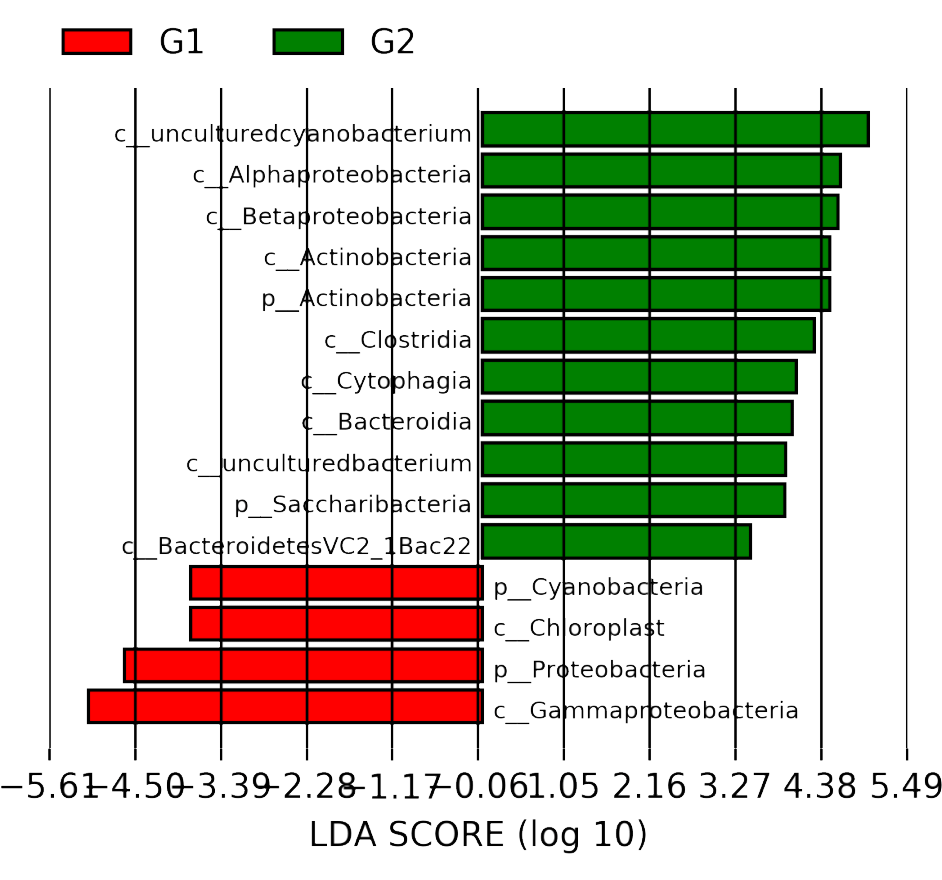
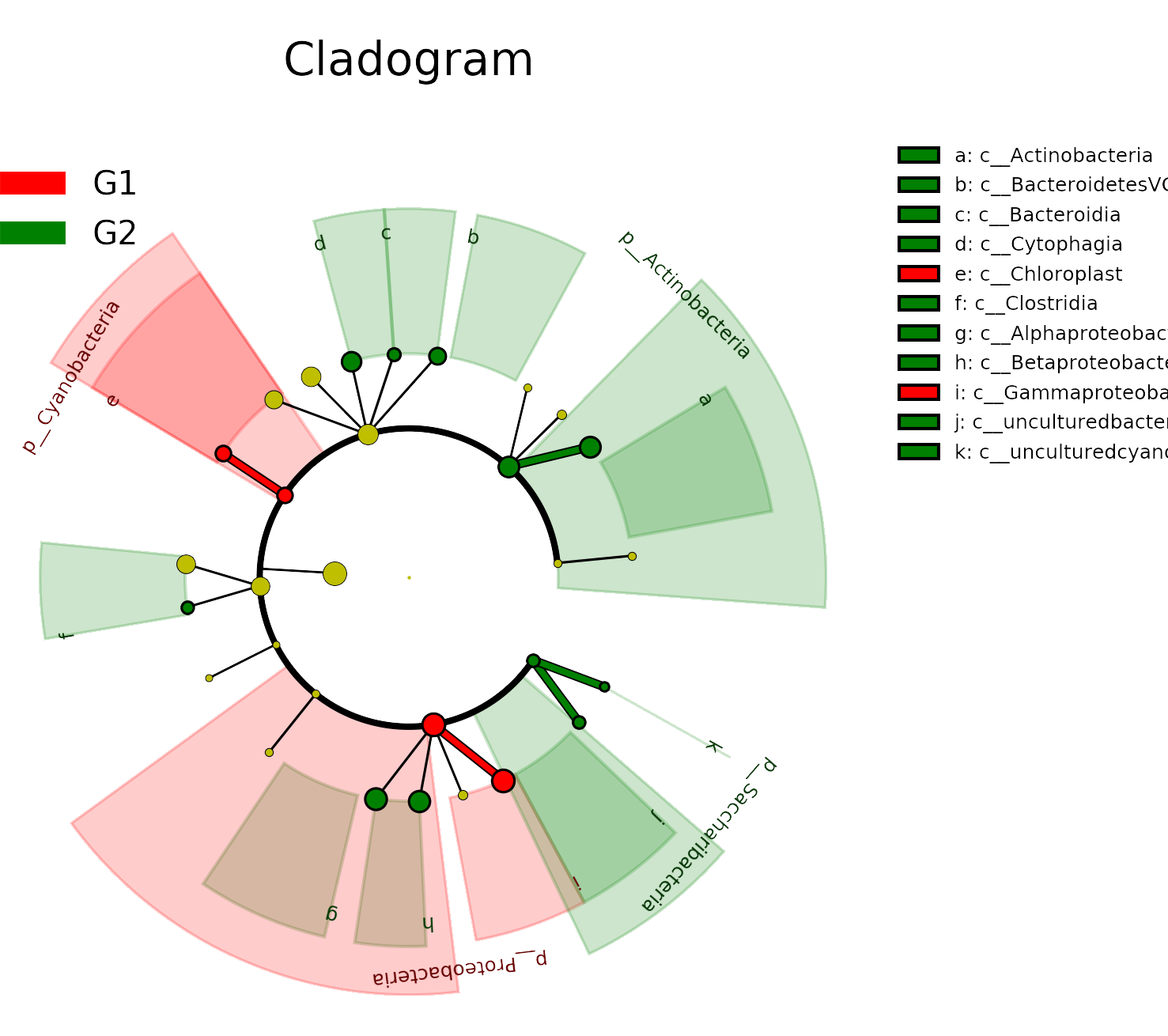
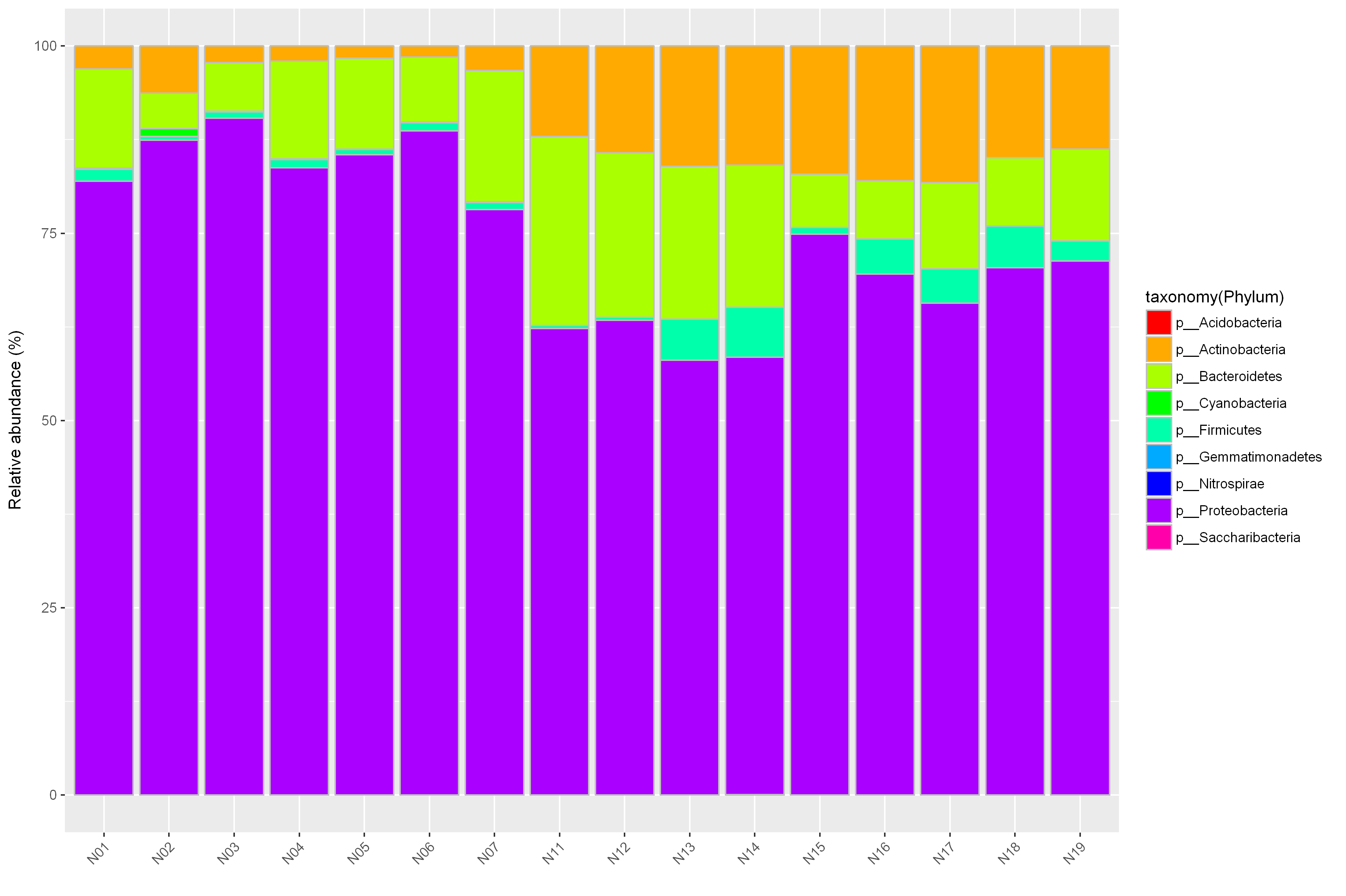
根据物种的相对丰度,选取门水平top5、属水平top15、种水平top20的优势物种,分析各样本间优势物种的分布情况。
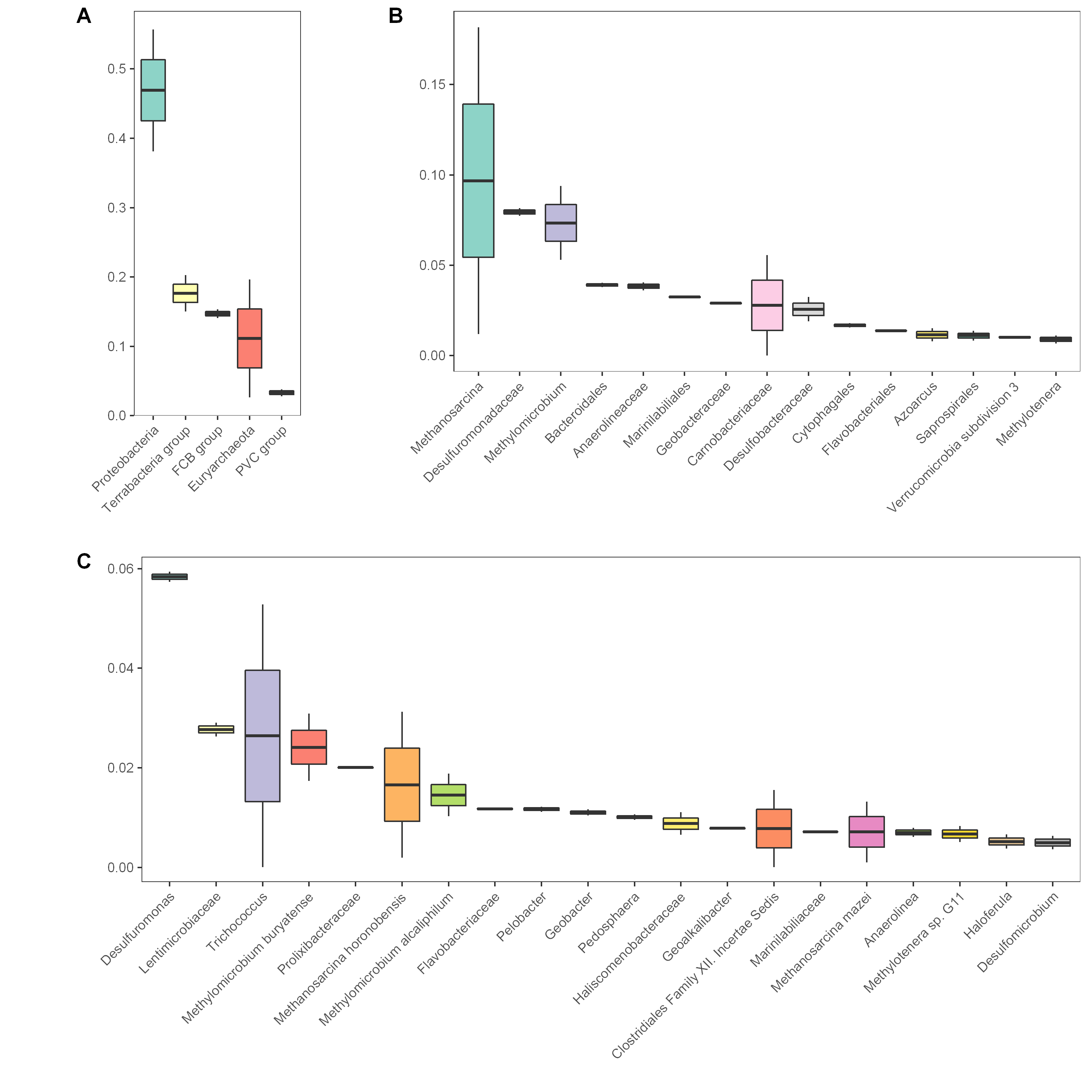
选定一个或多个需要分析的样品,选定一个分类学水平,按照相应多样性信息作图,反映各样品的群落结构。